Evaluation der Genauigkeit von Abständen aus NOE-Daten I
A. Kolmer, L. J. Edwards, I. Kuprov C. M. Thiele
Conformational analysis of small organic molecules using NOE and RDC data: A discussion of strychnine and α-methylene-γ-butyrolactone
J. Magn. Reson. 2015, 261, 101–109.
Die Extraktion von genauen Abständen aus NOE-Daten wird evaluiert. Dazu wird eine Auswertungssoftware entwickelt und die Ergebnisse mit RDC-Ergebnissen verglichen, um Stärken und Schwächen beider Methoden herauszufinden.
Die von Butts et al.1C. P. Butts, C. R. Jones, J. N. Harvey, Chem. Commun. 2011, 47, 1193–1195. an Strychnin 1 diskutierte Methode zur Extraktion genauer Abstände aus NOE-Daten enthält verschiedene Annahmen, die zu Über- oder Falschinterpretationen der Ergebnisse führen könnten. Es wäre daher möglich, dass die Schlussfolgerung von Butts et al., die experimentell bestimmten Abstände nur durch konformationelle Flexibilität im Siebenring erklären zu können, im Falle der Ungültigkeit dieser Annahmen eine Fehlinterpretation darstellt. Daher müssen diese Annahmen überprüft werden.

Die untersuchten Moleküle.
Gegen die Gültigkeit dieser Annahmen sprechen die folgenden möglichen Probleme oder Fehlerquellen. Die von Butts et al. verwendete Probe von Strychnin 1 wurde unter Sauerstoffatmosphäre abgeschmolzen. Allerdings gilt Sauerstoff als paramagnetischer Relaxationspartner, dessen Anwesenheit zu einer Veränderung der NOE-Intensitäten führen kann. Außerdem wurde ein einziger Datenpunkt bei einer Mischzeit von 500 ms aufgenommen, sodass die Gültigkeit der initial-rate-approximation nicht überprüft werden kann. Zusätzlich ist fraglich, ob die Wartezeit zwischen den Scans, die auf 1 s gesetzt wurde, ausreichend für komplette Relaxation ist bzw. ob trotz unterschiedlicher Relaxationszeitkonstanten ein „stationärer“ quantifizierbarer Zustand entsteht.
Neben diesen experimentellen Aspekten ist zu berücksichtigen, dass bei 600 MHz stark gekoppelte Protonen vorliegen. Trotz der Verwendung eines Moduls zur ZQS2M. J. Thrippleton, J. Keeler, Angew. Chem. Int. Ed. 2003, 42, 3938–3941. 3K. E. Cano, M. J. Thrippleton, J. Keeler, A. Shaka, J. Magn. Reson. 2004, 167, 291–297. 4M. J. Thrippleton, R. A. E. Edden, J. Keeler, J. Mag. Reson. 2005, 174, 97–109. ist es möglich, dass dieses abhängig von Kopplungskonstanten nicht effektiv arbeitet.5K. E. Cano, M. J. Thrippleton, J. Keeler, A. Shaka, J. Magn. Reson. 2004, 167, 291–297. Ein weiteres mögliches Artefakt entsteht durch Spindiffusion. Obwohl Spindiffusion nichtlinear sein sollte,6D. Neuhaus, M. P. Williamson, The Nuclear Overhauser Effect in Structural and Conformational Analysis, 2 Aufl., Wiley, Chichester, 2000. 7S. Macura, B. T. Farmer II, L. R. Brown, J. Magn. Reson. 1986, 70, 493–499. ist es möglich, dass diese Nichtlinearität durch Streuung der Datenpunkte nicht entdeckt wird.
Um diese möglichen Fehlerquellen zu untersuchen und auszuschließen, wurden Mischzeitserien von 50 – 400 ms in Schritten von 25 ms des 1D PFGSE NOE-Experiments8J. Stonehouse, P. Adell, J. Keeler, A. J. Shaka, J. Am. Chem. Soc. 1994, 116, 6037–6038. 9K. Stott, J. Stonehouse, J. Keeler, T.-L. Hwang, A. J. Shaka, J. Am. Chem. Soc. 1995, 117, 4199–4200. 10K. Stott, J. Keeler, Q. N. Van, A. J. Shaka, J. Magn. Reson. 1997, 125, 302–324. mit ZQS11M. J. Thrippleton, J. Keeler, Angew. Chem. Int. Ed. 2003, 42, 3938–3941. 12K. E. Cano, M. J. Thrippleton, J. Keeler, A. Shaka, J. Magn. Reson. 2004, 167, 291–297. 13M. J. Thrippleton, R. A. E. Edden, J. Keeler, J. Mag. Reson. 2005, 174, 97–109. aufgenommen und nach dem PANIC-Ansatz14S. Macura, B. T. Farmer II, L. R. Brown, J. Magn. Reson. 1986, 70, 493–499. 15H. Hu, K. Krishnamurthy, J. Magn. Reson. 2006, 182, 173–177. ausgewertet. Die Probe wurde unter Sauerstoffausschluss abgeschmolzen, und die Wartezeit zwischen den Scans wurde auf 15 s gesetzt (dies entspricht 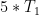
Der Einfluss der Nullquantenübergänge wurde durch Simulation von PANIC-Auftragungen mittels der Software SPINACH16Spinach version 1.3.1980, http://spindynamics.org/Spinach.php. 17H. J. Hogben, M. Krzystyniak, G. T. P. Charnock, P. Hore, I. Kuprov, J. Magn. Reson. 2011, 208, 179–194. 18I. Kuprov, J. Magn. Reson. 2011, 209, 31–38. 19A. Karabanov, I. Kuprov, G. T. P. Charnock, A. van der Drift, L. J. Edwards, W. Köckenberger, J. Chem. Phys. 2011, 135, 084106. getestet. Dazu wurden von Edwards die Möglichkeit konformationeller Flexibilität von Strychnin 1 sowie ein ZQS-Modul zur Verwendung in SPINACH entwickelt.
Dabei stellte sich heraus, dass das ZQS-Modul nicht immer perfekt arbeitet. Trotzdem stimmten die simulierten PANIC-Auftragungen nur dann mit den experimentellen überein, wenn konformationelle Flexibilität berücksichtigt wurde. Dies führt zur Schlussfolgerung, dass selbst wenn die bestimmten Abstände Nullquantenartefakte aufweisen, trotzdem konformationelle Flexibilität nötig ist, um die experimentellen Daten zu erklären. Da außerdem durch die Wahl der Größe des Basis-Sets auch Spindiffusion korrekt beschrieben werden kann, bedeutet dies analog, dass auch Spindiffusion keinen oder einen zu vernachlässigenden Einfluss auf den bestimmten Abstand hat.
Zur korrekten Beschreibung der Mittelung durch konformationelle Flexibilität bei der Auswertung von NOE-Daten wurde die Software WEEDHEAD entwickelt. Diese Software basiert auf dem Ansatz, dass durch Gleichung 1 gemittelte Abstände mit experimentellen Abständen verglichen werden sollen. Dazu werden Qualitätsfaktoren verwendet. Dabei wird keine Strukturoptimierung durchgeführt, stattdessen wird anhand der vorgegebenen Strukturvorschläge nach Minima der Konformerenpopulationen gesucht. Als Eingabedateien werden Strukturvorschläge als .xyz-Dateien sowie eine Datei mit den gemessenen Abständen benötigt.
Gleichung 1.

Mit Hilfe der bestimmten Abstände und der Software WEEDHEAD konnte ein Minimum des Qualitätsfaktors gefunden werden, welches der besten Übereinstimmung von gemittelten und experimentellen Daten entspricht. Dies liegt für Strychnin 1 bei 98% des Haupt- und 2% des Konformers mit alternativer Konformation des Siebenrings. Außerdem konnte die von Bifulco et al.20G. Bifulco, R. Riccio, G. E. Martin, A. V. Buevich, R. T. Williamson, Org. Lett. 2013, 15, 654–657. postulierte Flexibilität im Fünfring nicht beobachtet werden.
In der Literatur vorhandene RDC-Daten21C. M. Thiele, S. Berger, Org. Lett. 2003, 5, 705–708. 22C. M. Thiele, J. Org. Chem. 2004, 69, 7403–7413. 23 B. Luy, K. Kobzar, H. Kessler, Angew. Chem. 2004, 116, 1112–1115. 24N.-C. Meyer, A. Krupp, V. Schmidts, C. M. Thiele, M. Reggelin, Angew. Chem. 2012, 124, 8459–8463. 25N.-C. Meyer, Helikal chirale Polyacetylene in Katalyse und Analytik, Dissertation, TU Darmstadt, 2012. konnten mittels der MCST-Methode von RDC@hotFCHT26V. Schmidts, Entwicklung einer Auswertungssoftware zur Anwendung Residualer Dipolarer Kopplungen in der organischen Strukturaufklärung, Dissertation, TU Darmstadt, 2013. ebenfalls auf ihren Informationsgehalt bezüglich konformationeller Flexibilität des Siebenrings überprüft werden. Die Unterschiede der Qualitätsfaktoren sind allerdings nicht signifikant, sodass die RDC-Daten nur Hinweise auf Flexibilität beinhalten.
Nachdem die genaue Bestimmung von Distanzen aus NOE-Daten und deren Beschreibung implementiert ist, soll nun deren Robustheit geprüft werden. Dazu wird ein Molekül ausgewählt, das flexibler ist als Strychnin 1.
Für α-Methylen-γ-butyrolacton 2 existieren umfassende Studien zur Bestimmung konformationeller Flexibilität aus RDC-Daten.27C. M. Thiele, A. Marx, R. Berger, J. Fischer, M. Biel, A. Giannis, Angew. Chem. 2006, 118, 4566–4571. 28C. M. Thiele, V. Schmidts, B. Böttcher, I. Louzao, R. Berger, A. Maliniak, B. Stevensson, Angew. Chem. 2009, 121, 6836–6840. 29C. M. Thiele, A. Maliniak, B. Stevensson, J. Am. Chem. Soc. 2009, 131, 12878–12879. 30B. Böttcher, Studien zu Konformation und Dynamik eines Palladium-Allyl-Komplexes und eines Wirkstoffes mit residualen dipolaren Kopplungen, Dissertation, TU Darmstadt, 2011. Ursprünglich war die Konfiguration der beiden Stereozentren im flexiblen Fünfring unbekannt, diese konnte mit RDCs zu trans bestimmt werden.31C. M. Thiele, A. Marx, R. Berger, J. Fischer, M. Biel, A. Giannis, Angew. Chem. 2006, 118, 4566–4571. Die Population des Hauptkonformers wurde dabei per MCST zu 60%, per MCMT zu 65% und per Kopplungskonstantenanalyse zu 60% bestimmt.32C. M. Thiele, V. Schmidts, B. Böttcher, I. Louzao, R. Berger, A. Maliniak, B. Stevensson, Angew. Chem. 2009, 121, 6836–6840. 33C. M. Thiele, A. Maliniak, B. Stevensson, J. Am. Chem. Soc. 2009, 131, 12878–12879. 34B. Böttcher, Studien zu Konformation und Dynamik eines Palladium-Allyl-Komplexes und eines Wirkstoffes mit residualen dipolaren Kopplungen, Dissertation, TU Darmstadt, 2011.
Nun sollte anhand des α-Methylen-γ-butyrolactons 2 getestet werden, ob mittels des NOEs ebenfalls gleichzeitig Konfiguration und Konformation bestimmt werden können. Dazu wurden erneut quantitative NOE nach der an Strychnin 1 erprobten Methode bestimmt. Anschließend wurde die Software WEEDHEAD erweitert, damit Mittelungsvorgänge von Methylgruppen nach Gleichung 2 korrekt beschrieben werden. Die beste Übereinstimmung zwischen experimentellen und gemittelten Daten wurde für die trans-Konfiguration beim Konformerenverhältnis 67:33% gefunden.
Gleichung 2.
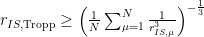
Dieses Ergebnis ist in exzellenter Übereinstimmung mit dem Ergebnis der RDC-Analyse. Es konnte deshalb gezeigt werden, dass genaue NOE extrahiert werden können. Diese können so wie RDC-Daten zur Konfigurations- und Konformationsanalyse genutzt werden. Im Falle von Strychnin 1 ist es durch die NOE-Analyse sogar möglich, Flexibilität zu detektieren, die anhand der RDC-Daten nicht eindeutig sichtbar ist. Um die Stärken beider Methoden auszunutzen, ist es daher empfehlenswert, beide Parameter komplementär zu verwenden.






