Potenziale von Industrie 4.0 im Großanlagenbau

Geschätzte Lesezeit: < 1 Minute.
T. Helmich, M. Artmeyer, A. Kolmer, T. Waldmann, O. Stecken
Potenziale von Industrie 4.0 im Großanlagenbau
Ergebnisse einer Gemeinschaftsstudie von maexpartners und der VDMA Arbeitsgemeinschaft Großanlagenbau






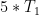 ). Auf diese Art konnte ein Großteil der Abstände von Butts et al. reproduziert und verfeinert werden.
). Auf diese Art konnte ein Großteil der Abstände von Butts et al. reproduziert und verfeinert werden.